
Tempo
CCS Research Pipeline for Whole-Genome and Whole-Exome Sequencing
Reproducible Workflows
Containerized workflows with Docker and Singularity
Portable
Tailored for LSF and AWS
User-Friendly
Written to be quickly run and used by anyone in the CMO
# Time-Efficient Mutational Profiling in Oncology (Tempo)
Tempo is a computational pipeline for processing data of paired-end whole-exome (WES) and whole-genome sequencing (WGS) of human cancer samples with matched normals. Its components are containerized and the pipeline runs on the Juno high-performance computing cluster (opens new window) at Memorial Sloan Kettering Cancer Center and on Amazon Web Services (AWS) (opens new window). The pipeline was written by members of the Center for Molecular Oncology (opens new window).
These pages contain instructions on how to run the Tempo pipeline. It also contains documentation on the bioinformatic components in the pipeline, some motivation for various parameter choices, plus an outline describing the reference resources used.
If there are any questions or comments, you are welcome to raise an issue (opens new window).
Note: Tempo currently only supports human samples. The pipeline has only been tested for exome and genome sequencing experiments, and all reference files are in build GRCh37 of the human genome.
# Table of Contents
# 1. Getting Started
# 1.1. Setup
# 1.2. Usage
# 1.3 Outputs
# 2. Pipeline contents
# 2.1. Bioinformatic Components
# 2.2. Reference Resources
- Genome Assembly
- Genomic Intervals
- RepeatMasker and Mappability Blacklist
- Preferred Transcript Isoforms
- Hotspot Annotation
- OncoKB Annotation
- gnomAD
- Panel of Normals for Exomes
# 2.3. Variant Annotation and Filtering
# 3. Help and Other Resources
# 4. Contributing
# 5. Acknowledgements
# Pipeline Flowchart
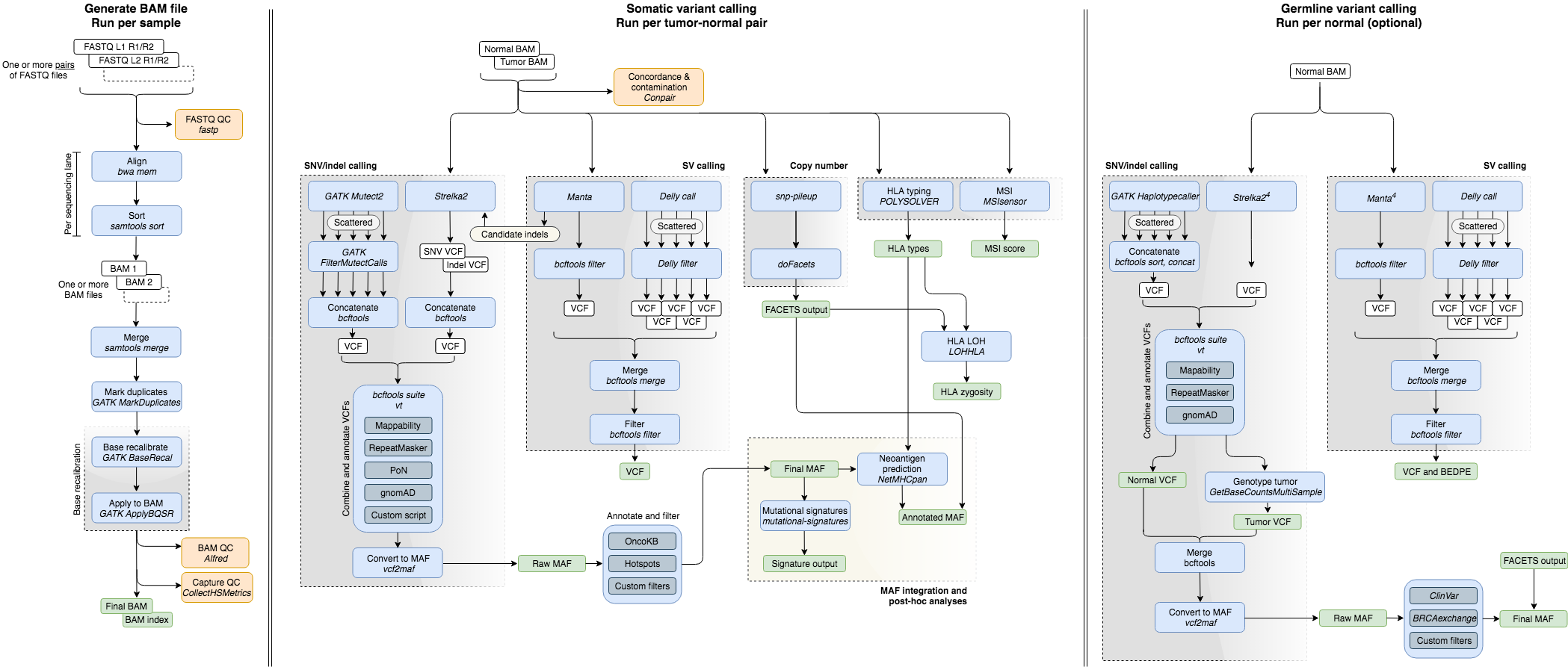
# Directed Acyclic Graph
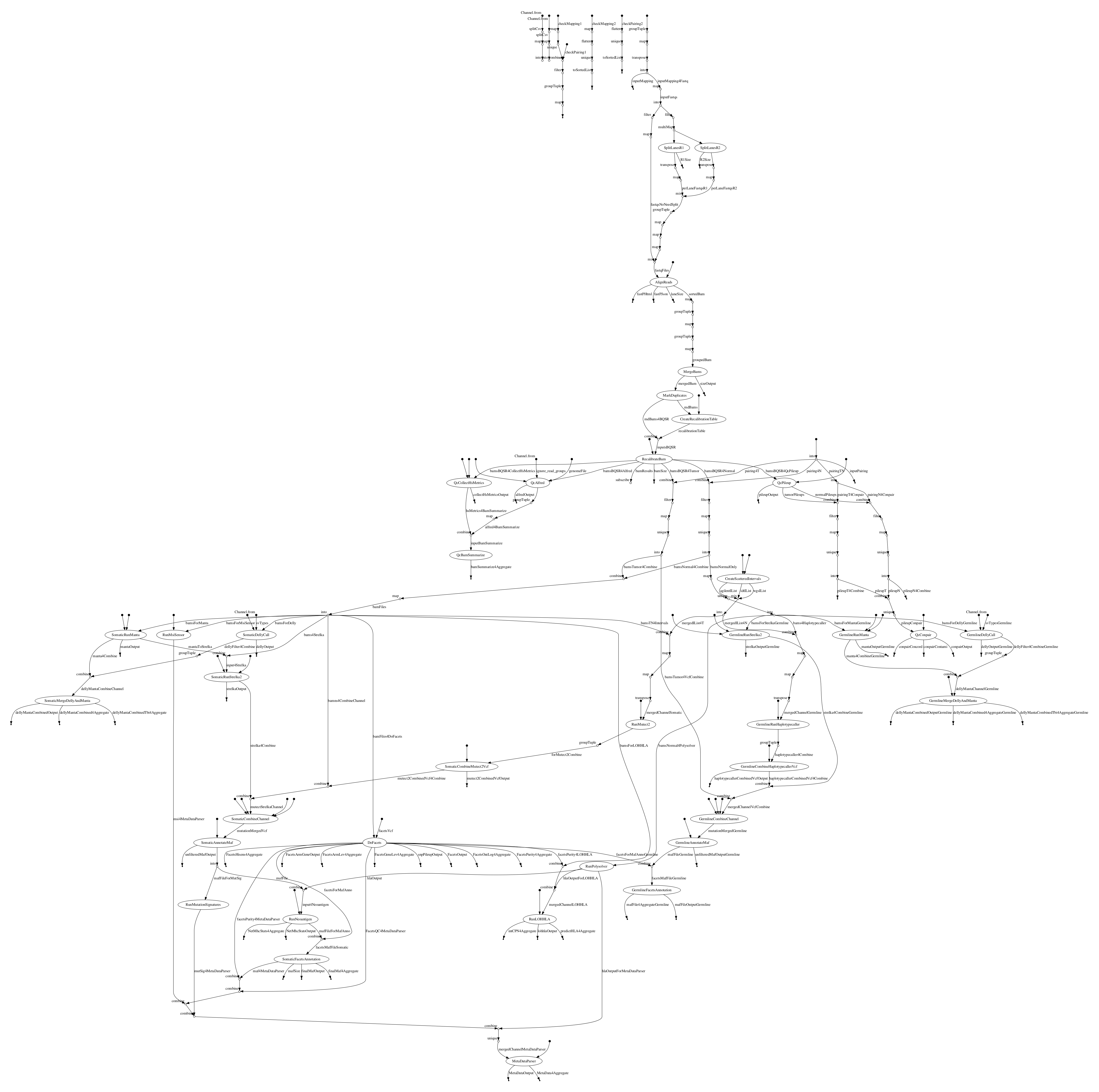
#
#